The concept of patient centricity is hardly new, but in recent months it has assumed new urgency as industry and regulators have had to adapt to an evolving landscape. The evolution is spurring substantial changes in how clinical trials are developed and implemented, as regulators revise their processes and issue updated guidance documents to encourage speed and efficiency while still ensuring patient safety.
The U.S. Food and Drug Administration (FDA) has been at the forefront of recent changes, notably through its issuance of Emergency Use Authorizations (EUAs) to facilitate the availability of testing kits, treatments, and vaccines; the establishment of its Coronavirus Treatment Acceleration Program; and the authorization of reference panel comparative data to enhance diagnostic capabilities. Over the past year and a half, the FDA has also issued 13 guidance documents, some of which have been updated to address various aspects of product development and clinical trials, reflecting a renewed focus on the patient journey in the wake of the pandemic (Table 1).
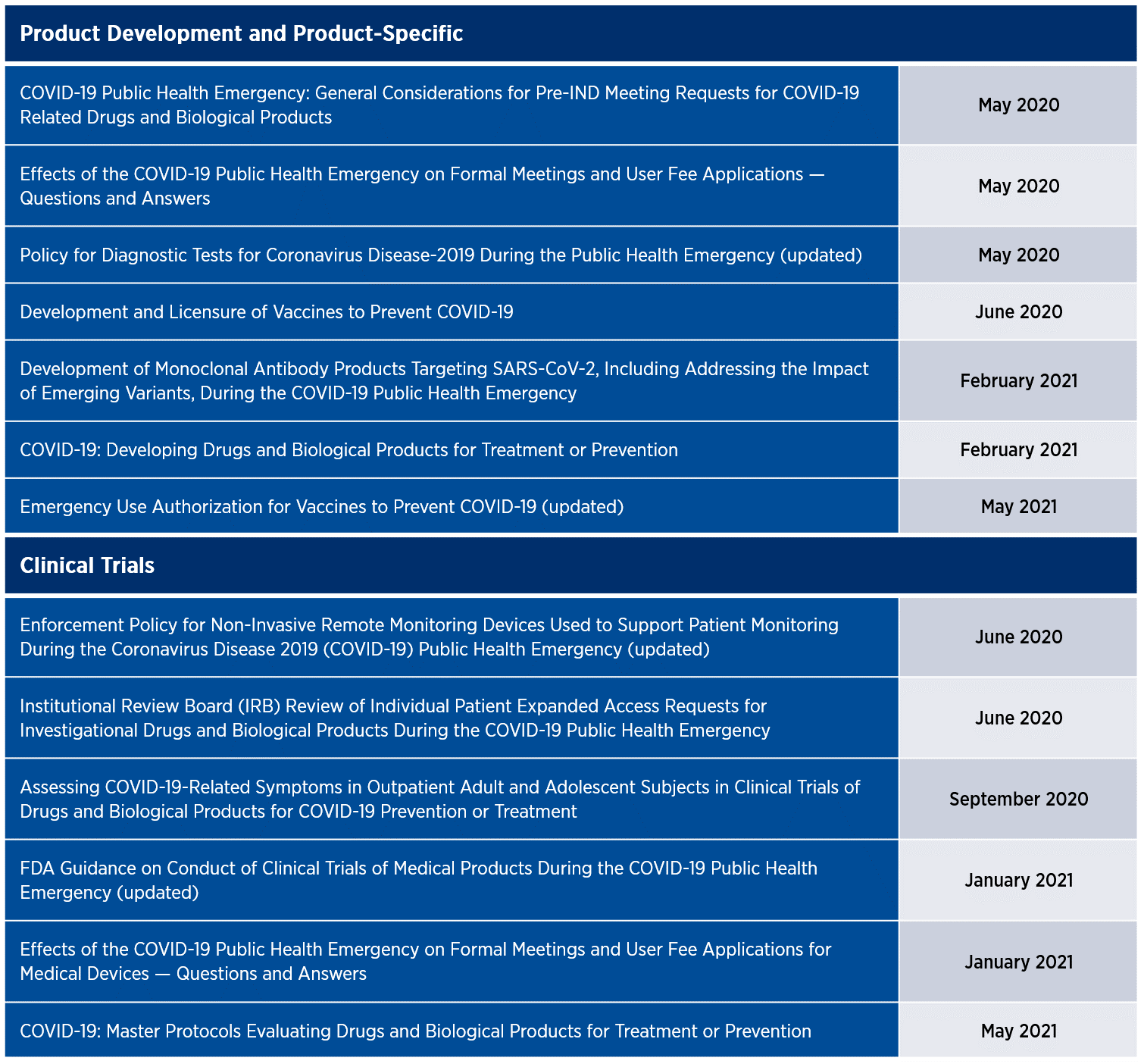
The FDA is encouraging the use of mechanisms such as pre-investigational new drug application (pre-IND) meetings, Q-submissions, and humanitarian device exemptions to expedite the review of new therapies and devices with potential efficacy and appropriate safety data to enter clinical trials. Those initiatives have helped compress review timelines, with some protocols reviewed within 24 hours and single-patient expanded access requests granted within three hours.
The European Medicines Agency (EMA) has exhibited a similar commitment to fast-tracking its review of clinical trial protocols for treatments and vaccines, shortening its standard 40- to 70-day timeline to approximately 20 days. The accelerated timeline includes pediatric investigation plans, which previously took up to 120 days to review. Additionally, the EMA has issued six updated guidance documents for clinical trials in the past year (Table 2), most of which aim to elevate patient-focused considerations that were gaining currency before the pandemic struck. On a parallel track, the EMA is increasingly implementing mechanisms such as rolling review, accelerated assessment, indication extensions (for already approved medicines), and compassionate use programs to expedite marketing authorization applications and to enhance patient access to investigational therapies.

The shortened review timelines and the uptick in trial approvals reflect not only the responsiveness and adaptability of the FDA, EMA, and other national competent authorities but also the creativity and inventiveness of the industry as all parties re-evaluate what the patient journey looks like in a shifting landscape. Whereas patient safety and trial integrity remain paramount, pandemic-related restrictions may necessitate further modification of certain processes as well as novel methods to engage and retain participants. As decentralized clinical trials (DCTs) eliminate the need for many participants to visit the investigational site, the shift to mobile health (mHealth), virtual visits, and electronic consent has accelerated. At the same time, participants lacking access to the investigational agents may need additional safety monitoring. Such adjustments may necessitate protocol amendments and coordination with institutional review boards (IRBs), ethics committees, and local authorities as appropriate.
The Future of Patient-focused Drug Development
The recent regulatory developments are expected to provide a new platform for future trials, reinforcing and amplifying the profile of clinical research participation in a post-COVID era. To that end, some regulations and processes will likely tighten to enhance patient safety, and others will loosen to make trial participation more convenient and accessible and simultaneously less burdensome. These changes will allow drug developers to adopt new methods of conducting trials more quickly and efficiently, particularly in terms of facilitating electronic data collection and patient monitoring for DCTs.
The updated regulatory guidelines may require overhauling good clinical practice principles and standard operating procedures (SOPs) to reflect the new trial landscape. In the near term, the updated guidelines and regulations will have an immediate impact on site initiation for new studies as sites implement newly mandated safety measures and other procedures to enhance the patient experience. Additionally, the shift to mHealth and remote monitoring technologies is expected to continue to accelerate, allowing more flexibility for trial participants.
In the longer term, sponsors and CROs will need to update their SOPs to comply with updated regulations and guidance documents designed to ease the patient journey. Some of the new regulations may require additional documentation and closer coordination with IRBs and ethics committees. While the industry will generally benefit from shorter approval timelines, trial sponsors and the CROs they work with will need to review their policies and strategies regularly. This will be especially important as the regulatory environment continues to evolve. From a patient recruitment perspective, this evolving environment seems poised to push the industry to more actively pursue greater diversity among clinical trial participants.
Balancing the Competing Imperatives for Speed and Safety
Intense competition has always been a defining feature of the clinical trial environment. The pandemic has disrupted research programs and development timelines across the industry, and trial sponsors are under increasing pressure to optimize their clinical development strategies and the patient experience. This is a balancing act that places patient safety at the forefront of every trial. As the evolving maze of new regulations intensifies the competing pressures for speed, efficiency, and safety, it is more important than ever for sponsors to consider patient needs at each step of the journey and to work with a partner with the expertise to guide them through today’s complex regulatory landscape.