


CAR-T cells are complex products, and translating them from basic and preclinical research to clinical trials and commercialization can be challenging. Understanding the regulations and developing a protocol that puts safety first—at every stage of development—are important first steps in bringing promising CAR-T cell therapies to the patients who need them most.
CAR-T Cell Therapies: Navigating Regulations and Managing Toxicities in Clinical Trials
Immuno-gene therapeutics are transforming the therapeutic landscape of cancer therapy. Last year’s approvals of the first two chimeric antigen receptor (CAR)-T cell therapies—tisagenlecleucel (marketed as Kymriah™) and axicabtagene ciloleucel (marketed as Yescarta™)—heralded a new era of precision oncology.
With more than 300 CAR-T cell clinical trials ongoing, we have seen only the tip of the iceberg.1 While CAR-T cell therapies have demonstrated dramatic efficacy in hematological malignancies, these treatments are associated with serious toxicities. As the number of clinical trials involving CAR-T cell therapies in both hematologic malignancies and solid tumors increases, there is an urgent need for sponsors of these trials—many of them small to midsize biotech or specialty pharma companies—to fully understand the regulations and safety considerations surrounding these novel treatments.
Understanding the Regulatory Landscape
Clinical trials of immuno-gene therapeutics are becoming increasingly common, and regulatory guidelines are steadily evolving to keep pace with advances in technology. As part of the study planning process, it is critical for sponsors of gene therapy technologies to gain a detailed understanding of the regulatory environment in every country where the study may be conducted.
FDA Guidance
The U.S. Food and Drug Administration (FDA) published its first guidance on gene therapy in 1998.2 Since then, the FDA has published numerous other guidance documents, including:
Preclinical Assessment of Investigational Cellular and Gene Therapy Products (November 2013)3: This guidance provides recommendations on the substance and scope of preclinical information needed to support clinical trials for investigational cellular therapies, gene therapies, and therapeutic vaccines.
Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products (June 2015)4: This guidance provides the FDA’s current thinking on clinical trials of investigational cellular therapy and gene therapy products in which the primary objectives are initial assessments of safety, tolerability, or feasibility of administration.
Recommendations for Microbial Vectors Used for Gene Therapy (September 2016)5: A supplement to the FDA’s Guidance for FDA Reviewers and Sponsors: Content and Review of Chemistry, Manufacturing, and Control Information for Human Gene Therapy Investigational New Drug Applications (INDs), this guidance offers recommendations concerning IND submissions for microbial vectors used for gene therapy in early-phase clinical trials.
Expedited Programs for Regenerative Medicine Therapies for Serious Conditions (November 2017)6: This document provides recommendations on the expedited development and review of regenerative medicine therapies for serious or life-threatening diseases or conditions. It also provides information on the provisions in the 21st Century Cures Act regarding use of the accelerated approval pathway for therapies that have been granted the regenerative medicine advanced therapy (RMAT) designation.
Evaluation of Devices Used with Regenerative Medicine Advanced Therapies (November 2017)7: This draft guidance provides the FDA’s recommendations regarding evaluation of devices used in the recovery, isolation, or delivery of RMATs.
Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use (updated December 2017)8: This guidance clarifies the definitions of minimal manipulation and homologous use and facilitates understanding of how regulatory criteria apply to human cells, tissues, and cellular and tissue-based products.
EMA Guidance
The latest guidance from the European Medicines Agency on anti-cancer medicinal products provides the following guidelines on immuno-modulating products:9
- Non-clinical in vitro and in vivo proof-of-concept studies are needed to justify the planned starting dose and schedule in Phase I studies.
- Rationale for the starting dose may be supported by using the Minimal Anticipated Biological Effect Level (MABEL) approach, or by non-clinical and clinical data from related compounds.
- Information on the differential expression of the target antigen in human tumor and healthy tissues should be provided.
- In cases where no relevant and predictive animal model is available, in vitro studies with human cells (e.g., in vitro T-cell priming assays) might be suitable.
- The aim of early clinical trials is to determine safety, as well as the dose and schedule that induce a desired immune response.
- Dose-finding studies are generally required to establish the recommended Phase II dose.
- Monitoring the immune response (i.e., the induction of antigen-specific T cells or the presence of a humoral response) is of interest for determining appropriate dose and schedule.
More recently, the EMA published a concept paper on the revision of its guideline on quality, non-clinical, and clinical aspects of medicinal products containing genetically modified cells, which originally went into effect in 2012.10 This concept paper calls for a multidisciplinary revision of the existing guidelines to reflect current technology and experience, as well as to provide specific guidance for the development of CAR-T cells and related products. It is anticipated that a draft revised guideline will be available in 2018.
For global studies, it is important to remember that the EU comprises many countries, and the regulatory environment in certain countries may require additional procedures outside of standard regulatory and ethical review. For example, in some EU member states, CAR-T cells are classified as genetically modified organisms and may require an environmental risk assessment for clinical trial approval.11 Certain EU member states may also require sponsors to notify other government bodies when a clinical trial of an immuno-oncology agent is being conducted.
Sponsors should also invest time in understanding the difference between the contained use (Directive 2009/31/EC) and deliberate release (Directive 2001/18/EC) risk classifications for genetically modified organisms, and how each EU member state regards clinical trials with immuno-gene therapeutics. Additional rationale and scientific documents, such as an environment risk assessment or summary notification information format, may be required.
Putting Safety First in CAR-T Clinical Trials
Anticipating, preventing, and managing toxicity is a critical component of clinical studies involving CAR-T cells.
Safety Considerations with CAR-T Cell Therapies
The adverse events (AEs) associated with immuno-oncology agents such as CAR-T cells differ from those associated with cytotoxic therapies. In addition, the AEs following T cell-based therapies vary widely in severity and time of onset. In some cases, the toxicities may persist for the lifespan of the genetically modified T cell.12
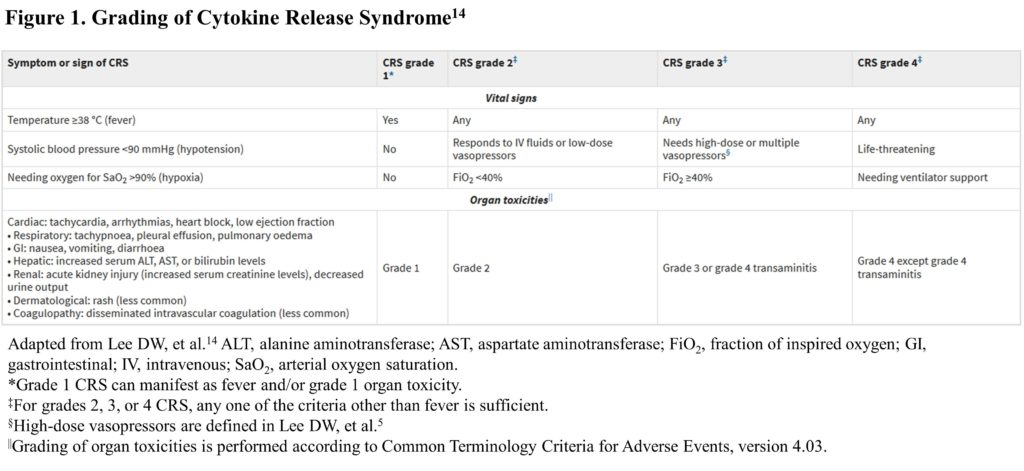
To date, cytokine release syndrome (CRS) is the most prevalent AE following CAR-T cell infusion. CRS can range in severity from low-grade constitutional symptoms to life-threatening multi-organ dysfunction. In rare instances, CRS can evolve into hemophagocytic lymphohistiocytosis, and it has been linked to two reported deaths in clinical trials.13 The second most common AE is CAR-T cell-related encephalopathy syndrome (CRES). Combination studies of CAR-T cell therapies with other immuno-oncology therapies could potentially increase toxicity in terms of severity and range of AEs.
 Designing a CAR-T Study Protocol
Designing a CAR-T Study Protocol
Safety considerations related to CAR-T cells may impact both trial design and trial management.
Therefore, the following criteria should be clearly defined in the study protocol:
- Prophylactic measures
- Management and dose adjustments of other medications
- Permissible concomitant medications and supportive care
- Any immediately reportable AEs
- What does not need to be reported; e.g., progression of disease
It is also important to consider other factors that may impact the safety profile, including:
- Concomitant medications
- Disease being studied, prior lines of therapy, and disease burden
- CAR design and production and amplification of the CAR-T cell
- Patient preconditioning and administered doses
Regular safety review meetings and data safety and monitoring are required for any study involving CAR-T cell therapy, and adjudication of dose-limiting toxicities may be considered.
Managing Toxicity with CAR-T Cell Therapy
Advancements in the development of CAR-T cell therapies, such as integration of genetic constructs containing “safety (suicide) switches” or “elimination genes” and “remote-controlled” CARs, may help to limit toxicities. Suicide switches and elimination genes are designed to eliminate target CAR-T cells when life-threatening toxicities develop. Remote-controlled CARs include an inducible gene-regulatory system that enables controlled expression.
Recently, the CAR-T-cell-therapy-associated TOXicity (CARTOX) Working Group developed a set of recommendations for monitoring, grading, and managing toxicities in patients treated with CAR-T therapy, and these recommendations should inform a patient’s schedule within a protocol design.14
These recommendations suggest the following procedures before and during CAR-T cell infusion:4
- Baseline brain MRI to rule out any central nervous system disease
- Central venous access
- Cardiac monitoring by telemetry beginning on the day of CAR-T cell infusion and continuing until CRS resolves
- Tumor lysis precautions for patients with bulky tumors
- Consideration of seizure prophylaxis for therapies known to cause CRES
- Hospitalization for at least seven days after CAR-T cell infusion
The protocol for patient monitoring after CAR-T cell infusion should include:4
- Vital signs every four hours, along with close monitoring of oral and intravenous fluid input and urine output and daily measurement of body weight
- Daily review and physical examination
- Daily blood counts, complete metabolic profiling, and coagulation profiling
- Daily C-reactive protein and ferritin levels
- Assessment and grading of CRS twice a day, and whenever the patient’s status changes
- Maintenance intravenous fluids with normal saline to ensure adequate hydration
- Assessment and grading of CRES using the CAR-TOX 10-point neurological assessment (CARTOX-10) every eight hours
CARTOX-10 involves asking a patient to:4
- Name the year, month, city, hospital, and president or prime minister of their home country (5 points)
- Name three nearby objects (3 points)
- Write a standard sentence (1 point)
- Count backward from 100 by tens (1 point)
The entire care team and investigative site staff need to be trained to recognize the unique toxicities of CAR-T cell therapies and respond accordingly and in a timely fashion. Accurate assessment and appropriate management of toxicities can help mitigate adverse outcomes, maximizing the benefit of adoptive T cell therapies while minimizing the risk of life-threatening complications.
Compiling a timely submission for an Investigational New Drug Application or Biologics License Application requires an integrated, multifaceted clinical development team in which cross-fertilization is paramount. It is imperative that a gap analysis be conducted at the outset to help navigate the nuances of the global regulatory environment and the toxicity of immuno-modulating products and gene therapy products such as CAR-T therapy.
References:
1. ClinicalTrials.gov. Accessed July 1, 2018.
2. U.S. Food and Drug Administration. Guidance for Industry: Guidance for Human Somatic Cell Therapy and Gene Therapy. Available at https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM081670.pdf.
3. U.S. Food and Drug Administration. Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products. Available at https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM376521.pdf.
4. U.S. Food and Drug Administration. Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products: Guidance for Industry. Available at https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM564952.pdf.
5. U.S. Food and Drug Administration. Recommendations for Microbial Vectors Used for Gene Therapy: Guidance for Industry. Available at https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM466625.pdf.
6. U.S. Food and Drug Administration. Expedited Programs for Regenerative Medicine Therapies for Serious Conditions: Draft Guidance for Industry. Available at https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM585414.pdf.
7. U.S. Food and Drug Administration. Evaluation of Devices Used with Regenerative Medicine Advanced Therapies: Draft Guidance for Industry. Available at https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM585417.pdf.
8. U.S. Food and Drug Administration. Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use. Available at https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM585403.pdf.
9. European Medicines Agency. Guideline on the evaluation of anticancer medicinal products in man. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/11/WC500238764.pdf.
10. European Medicines Agency. Concept paper on the revision of the Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/07/WC500231995.pdf.
11. Hartmann J, et al. “Clinical Development of CAR T-cells — Challenges and Opportunities in Translating Novel Innovative Treatment Concepts.” EMBO Mol med 2017;9(9):1183-1197.
12. Bonifant CL, et al. “Toxicity and Management in CAR T-cell Therapy.” Mol Ther Oncolytics 2016;2:16011.
13. Hartmann J, et al. “Clinical Development of CAR T-cells — Challenges and Opportunities in Translating Novel Innovative Treatment Concepts.” EMBO Mol med 2017;9(9):1183-1197.
14. Lee DW, et al. “Current Concepts in the Diagnosis and Management of Cytokine Release Syndrome.” Blood 2014;124:188-195.
15. Neelapu SS, et al. “Toxicity Management After Chimeric Antigen Receptor T cell Therapy: One Size Does Not Fit All.” Nat Rev Clin Oncol 2018 Feb 13. Epub 2018 Feb 13.


